


- 更新时间:2024-06-11 09:05
所属行业:咨询 管理咨询 -
发货地址:上海金山
信息编号:296565524,公司编号:16129072
产品规格:不限
产品数量:9999.00 个
包装说明:不限
产品单价:面议
官方网址:http://sungoyuan.b2b168.com/
- 13818104617 袁玲
- 留言数:24人
- 查看数:2人
行走机器人出口EU2017/745认证要求 医疗器械新MDR认证知识
- 相关产品:
- 所在区域:上海浦东陆家嘴
- 经营性质:外商独资企业
- 企业类型:商业服务
- 注册地:上海市
- 主营产品:FDA510K认证,欧盟自由销售证书,MDR认证,ISO13485认证,MHRA注册,英国授权代表,UKCA认证,瑞士代表,CH,REP,FDA验厂辅导
- 企业已认证
- 个人实名已认证
- 产品分类
- 商家其他产品推荐
- 产品详细信息
其主要过程包括:确定根据指令的定义,产品是否属于器械,哪个欧盟指令适用于所考虑的器械:器械指令(93/42 / EEC),体外诊断器械指令(98/79/EC)或有源植入式器械指令(90/385 / EEC)
确定器械的分类。
实施质量管理体系(如适用)。大多数公司使用ISO 13485来满足要求。
MDR法规 共10 章123 条,并附有17 个附录。
在产品上市之前即明确了上市后制造商的责任和义务,并使得上市后有相应的依据。其次是持续的理念,关于评估,法规规定:公告机构应至少每隔12 月开展一次适当的审核和评估,以确保相关制造商采用批准的质量管理体系和上市后计划,公告机构至少应每隔五年随机对制造商进行一次现场突击审核。包括对制造商经营场所的审核,必要时还包括对制造商的供应商和/ 或分包商的审核。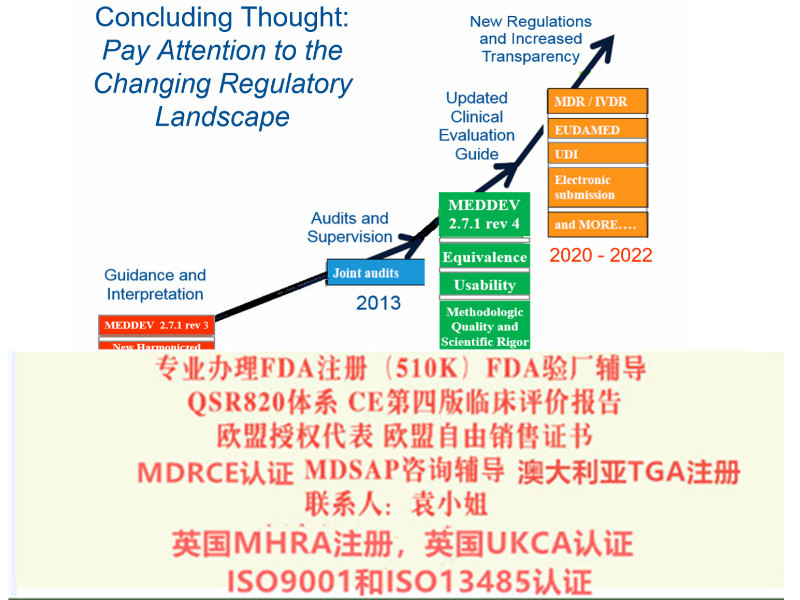
MDR的主要变化 1.扩大了应用范围 2.提出了新的概念和器械的定义 3.细化了器械的分类 4.完善了器械的通用和性能要求 5.加强对技术文件的要求 6.加强器械上市后的 7.完善评价相关要求 8.提出Eudamed数据库的建立和使用 9.提出器械的可追溯性(UDI) 10.对NB提出严格的要求在新版MDR 2017/745/EU中,更是完善了评估(包括器械售后追踪)和调查的执行、评估、报告和更新资料的相关要求。对特定III类和IIb类器械,评估报告中要考虑咨询小组的意见;对植入式和III类器械,提出考虑研究;要求评估报告按照售后追踪所取得的数据进行更新;
在上市后要求中,经济运营商同时负有相应的责任和义务。法规对各方义务的描述更为明确也更为具体,对于制造商的要求更为细化,因此新法规执行后,各方应先明确自身职责和义务,规范有序地开展生产和市场活动,应审核确认上游供应商是否符合规定,并确认能够自己的下游流程符合规定,应按照对应的警戒系统的要求进行或配合事件上报,配合完成现场纠正措施,并依据职责组织培训。法规中规定了对于一次性使用器械的再处理即复用的要求。
瑞士也已经不认可欧洲的CE认证,您有产品出口瑞士吗?是否有做瑞士代表以及瑞士注册的?
- 商家联系方式
- 我要给商家留言
- 商家产品推荐










- 相关产品推荐
-
架空乘人矿山井下可摘挂式猴车抱索器K9型 索道悬挂式吊椅用连接件
赤峰耐腐蚀塑料管
电动病床的美国FDA510K认证 510K申请需要什么资料
江阴国标工业异丁醇 可溶于水 沸点为107.89 °C
瓦房店到井陉搬家公司 安心送货_贴心服务
尿道扩张器出口自由销售证书认证怎么申请 什么是自由销售证书
火花直读光谱仪上市公司 光谱仪 罗兰圆
衡阳1FL6044-2AF21-1AG1伺服电机 具有多种控制模式和接口
青岛聚丙烯增强管
黑龙江判官佛像
淮安大量西门子触摸屏回收 资源再生
厂家供应 河*黑天土神神像厂家
张家港饮料生产线设备-酸奶全自动饮料三合一灌装机
苏州加固设计施工电话 施工*
长安区印刷经营许可证申请条件 拓展市场
DC96V4.5KW直流无刷电机驱动器- DC96V4.5KW直流无刷电机控制器
碳硫联合测定仪 碳硫分析室
钢铁厂风管机油站不锈钢滤芯1.1000H10XL-A00-0-M
银川智能旗杆升降控制系统厂家 抗风防锈 四爪抓地更稳固
漯河大蒜油厂家 植物精油 精细工艺
平凉市过河管道铺设安装公司-水下钢围堰切割拆除队伍