


- 更新时间:2024-06-04 10:09
所属行业:商务服务 认证服务 -
发货地址:上海金山
信息编号:296074988,公司编号:14235332
产品规格:不限
产品数量:2345667.00 个
包装说明:不限
产品单价:面议
官方网址:http://sungofda.cn.b2b168.com/
- 13818104617 袁小姐
- 留言数:12人
- 查看数:5人
轮椅CFS自由贸易证书 需要交年费嘛
- 相关产品:
- 所在区域:上海金山石化
- 经营性质:外商独资企业
- 企业类型:商业服务
- 注册地:上海市金山区
- 主营产品:CE,MDR认证,MDR,CE认证,IVDR,欧代,EU2017/745认证,2017/746,EC,REP,CE技术文件,CE第四版临床评估报告.SUNGO,EN,455,UK,EN12184,清洗验证,FDA验厂
- 企业已认证
- 个人实名未认证
- 产品分类
-
FDA验厂
CE第四版临床评价报告
MHRA注册/欧盟授权代表
欧盟自由销售证书
医疗器械单一体系审核MDSAP
MDR CE认证
欧盟授权代表
US Agent,FDA美国代
美国FDA注册/认证
MDR EU2017/745认证
MDEL注册
- 商家其他产品推荐
-
棉球瑞代 SUNGO的欧盟授权代表 EC REP怎么申请
拐杖的MDR CEMDR EU 2017-745认证 欧盟医疗器械CE认证之MDR 2017/745 EU 完整版
床头靠背MHRA注册 MHRA注册 mhra如何申请
一次性使用电子膀胱镜怎么申请欧盟自由销售证书 哥伦比亚要欧盟自由销售证书FSC 和MHRA自由销售证书区别
浴室扶手质量管理体系认证 要求是什么
胰岛素注射针质量管理体系认证 iso13485医疗器械体系认证 要求是什么
椎间融合器美国FDA注册 医疗器械FDA的注册步骤
电动病床FDA注册 美国fda注册 医疗器械FDA注册步骤
软性亲水接触隐形眼镜美国FDA注册 fda医疗器材注册 医疗器械FDA注册步骤
注射器MHRA认证 申请流程介绍
- 产品详细信息
SUNGO至今为客户申请了数千份自由销售证书,其签发机构包括了国内行业协会、国内主管机构和欧盟主管机构,其中欧盟主管机构(英国和荷兰)签发的占到90%以上。
我公司办理产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德等CE认证(MDD/MDR法规),CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
用品才可以办理欧盟自由销售证书的,欧盟国家有CE证书,ISO 13485证书就可以了, 中东,南美 尤其是:沙特、阿根廷、埃及 这些国家会要这种欧盟自由销售证书的,客户销售企业的产品的时候,当地要求必须注册成功才可以销售产品,那么注册的时候是需要这些文件的,MHRA颁发的自由销售证书,能企业生产的产品满足欧盟的法规要求,可以在欧盟市场自由销售。但通常欧盟国家不会要求企业出具CFS,只需CE证书,即可完成清关。
欧盟成员国以外的一些国家,比如埃及、巴西、阿根廷、印度尼西亚、委内瑞拉等国家会要求企业出示CFS证书。
器械CE认证(MDD 93/42/EEC)
体外诊断器械CE认证(IVDD 98/79/EC)
认证步骤:
1. 依据企业提供的产品信息,确定产品的分类并为企业特定要求制定认证方案。包括适用欧盟指令标准分析、对于产品认证模式分析、认证时间安排等;
2. 选择符合性评估途径
3. 编制技术文件 TECHNICAL CONSTRUCTION FILES 包括基本要求检查表E.R.C、产品风险分析报告、符合性申明、产品标签设计、产品说明书的修订等。
4. CE符合性声明 EC Declaration of Conformity
5. 委任欧盟授权代表European representative
6. 由欧盟授权代表将制造商及产品在欧盟主管机关注册
7. 质量体系的建立和维持
8. 建立售后警戒系统/加贴CE标签并将产品投放市场
欧盟自由销售证书Free Sales Certificate
欧盟自由销售证书也叫出口销售书 英文名称为:Free Sales Certificate、Certificate of Free Sale或者Certificate For Exportation of Medical Products;简称:FSC 或 CFS。
自由销售证书源于欧洲, 起初,欧洲经济区协定(EEA)个别成员国以及成员国境内的民间协会机构为了促销本国的产品到EEA的第三国,为当地制造商出具自由销售证书,其内容是是产品满足相关国家标准,满足相关的指令要求,产品*可靠,质量达到相关要求,可以在本国本地范围内自由销售,并允许出口之类的。初的自由销售证书由欧洲民间协会、商会等机构出具,后经欧洲一些国家完善形成一套适合自己国家的体系。源于欧洲的这种自由销售贸易壁垒,逐渐被世界部分,他们在进口产品时会要求货物发货方提供相应的自由销售证书。
欧盟授权代表:为了更好地维护欧盟的顾客和环境,为了完成产品的可追溯性(Traceability),欧盟的法规要求,制造商投放到欧盟市场的加贴了CE标志的产品有必要标有制造商的名称和联络地址;假设制造商来自欧洲经济区EEA(包含EU与EFTA)以外的国度,其产品有必要一同标有制造商和欧盟授权代表的名称和联络地址。
欧盟会为了进步全体的市场功率,请求一切成员国都应称心低法令请求,欧盟会和海关协作,并与相关方(制造商、欧盟受权代表、进口商、分销商)展开协作,树立完善的产品追溯系统。欧盟会首要关注高风险范畴,例如:器械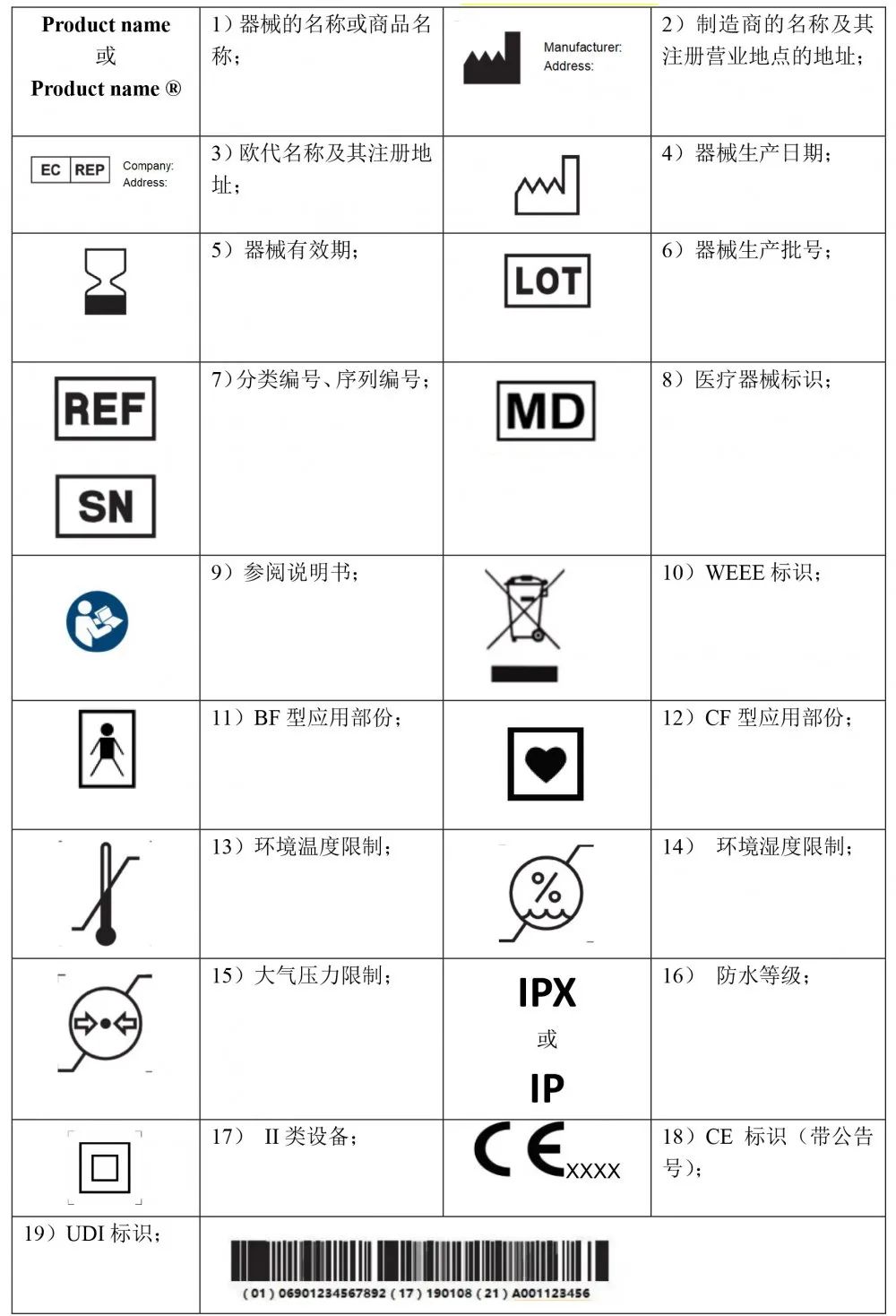
2017 年4 月5 日,欧洲议会和理事会正式签发了欧盟关于器械第2017/745 号法规(MDR,EU2017/745),5月5日,欧盟(Official Journal of the EuropeanUnion) 正式发布该法规。2017 年5 月25 日,MDR 正式生效, 替代了原器械指令(MDD,93/42/EEC)和主动植入式器械指令(AIMD,90/385/EEC)。
MDR 由指令升级为法规,提高了对欧盟成员国的约束力,具有直接约束性,*各国转化为本国的法律法规的形式即可落实实施。内容上,MDR 在整合原指令的基础上,大幅提升了有关器械认证的规范和限制,例如关于产品分类规则、器械的可追溯性、性能研究的规范、增加上市后的产品*性和有效性的等方面。MDR 共10 章123 条,并附有17 个附录。
一、关于法规过渡期
MDR 过渡期为3 年,共涉及四个时间点(见表1):
欧盟器械新法规MDR主要变化情况介绍
仅具有根据90/385/EEC 和93/42/EEC 指令签发的证书的器械可投放市场的前提是自MDR 适用之日起,其在设计和预期目的上无显著变化并符合新法规有关市场后监察、市场监察、警戒、经济运营商及器械注册的规定。
通过豁免指令的形式上市,且符合新法规的器械可在2020 年5 月26 日之前投放市场。并可在2020 年5 月26 日前并通知符合新法规的符合性评估机构。公告机构可在2020 年5 月26 日前, 采用合规的符合性评估流程并按照新法规签发证书。
我公司办理产品出口欧盟、美国、中东南美等国家的各种认证:
TUV莱茵,TUV南德等CE认证(MDD/MDR法规),CE技术文件编订, CE第四版评价(MEDDEV 2.7.1 Rev 4)编写,欧盟授权代表,欧盟自由销售证书,ISO13485:二016,器械单一审核程序(MDSAP)审核、美国FDA注册(含FDA510K申请),FDA美国代理人服务/ FDA QSR820验厂及整改,FDA警告信应对&RED LIST REMOVAL/QSR820体系/OTC验厂及整改,英国BRC认证咨询,BSCI验厂;口罩NELNSON(尼尔森)检测:(EN 14683检测、BFE检测,VFE检测、PFE检测)、手术衣EN13795测试、防护服type5/6 (PPE认证咨询(89/686/EC个人防护指令)。
对于珠三角地区,即广州、深圳、佛山、东莞、惠州、中山、珠海、江门、肇庆,加上新规划扩容汕尾(深汕特别合作区)、清远、云浮、河源、韶关,许多企业进行生产销售。
平日里,总是有人会问,什么是自由销售证,埃及、阿根廷、哥伦比亚、越南、沙特等客中提到的欧盟自由销售证是什么,怎么办,流程复杂吗,需要提供什么材料,有效期多长,是否是有效的等等的问题。
的确在如今的贸易往来中,各国也会对产品在其本国市场上的顺利上市及流通设置一些认证关卡,也就是一些认证注册的技术壁垒需要打破。尤其是器械类产品,如根管锉根管马达等器械、器输液器耗材、敷料创可贴等,多国当局会要求其在当地、局注册后才能上市。
产品在欧盟销售,只要有CE证书即可。销售到非欧盟国家,很多国家会要求您提供欧盟主管当局签发的自由销售证书。
- 商家联系方式
- 我要给商家留言
- 商家产品推荐



- 相关产品推荐
-
国产代替压力变送器校准仪 压力传感器 性能稳定
广州智能门锁日本TELEC认证办理标准
厦门兽药洁净度检测报告 国内GMP认证机构 广州市微生物研究所
深圳手机电子产品包装盒定制 电子包装
贵港房屋质量鉴定 营业执照需房屋*检测报告 行业经验丰富
解放劳动力 煤矿罐笼无线定位充电装置 智能型
临沂CPU 1515SP PC2 T
金华手术室净化工程厂家 品质优良
能降低后工序处理难度 3M 广州钢纸磨片单价
海口GMP认证报告 兽药生产车间检测 微生物研究所
河北电动轮椅EN12184检测 手动轮椅FDA510K 流程介绍
福州EVA扬子石化厂家 高弹力发泡
井下全自动液压风门 平衡式无压风门 红外线感应灵敏
满焊制作牢固结实 井下全自动平衡风门
成都到汉中电梯运输 托运公司物流
黄山手术室净化安装价格 品质优良
合肥智能空降闸图片 厂家销售
郫县幼儿园灭蟑螂服务
河北二手蒸发器电话 二手浓缩蒸发器
儿童发育行为评估量表 黑龙江儿童心理行为发育量表软件
不炸的黄铜氩弧焊丝制造商 204SM 焊接紫铜