

2023年年末,FDA发布了“510(k) Third Party Review Program and Third Party Emergency Use “510(k)第三方审核计划和第三方紧急使用授权(EUA)审核指南草案。
该草案概述了FDA目前对510(k)第三方审核计划和紧急使用授权(EUA)请求的关键想法,描述了FDA对第三方审核510(k)提交和第三方审核组织对EUA请求的期望,并提供了有关510(k)第三方审核计划的最新建议。相关内容曾在 2020 年最终指南“510(k) Third Party Review Program ”510(k)第三方审核计划中讨论过。
相较于之前发布的最终指南,本次更新主要增添了对510(K)第三方审核计划和EUA第三方审核之前的描述及区分等内容。
01什么是510(K)第三方审核
510(k)第三方审核计划是一种自愿的医疗器械替代审核流程,即允许经认可的第三方审核组织(3P 510k审核组织)审核某些中低风险的医疗器械。
该计划旨在加快中低风险器械510(k)审核速度,让FDA可以将资源集中在高风险器械的审核上,但FDA仍保持对符合第三方审核资格的中低风险器械的审核监督权限。
FDA第三方510(k)审核基本流程图如下:
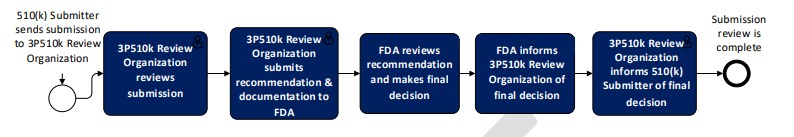 按上图所示,510(k)申请人首先向510(k)第三方审核机构递交技术文件,第三方审核机构按照与FDA相同的标准审核完成后向FDA递交推荐意见,FDA审核之后做出最终决定并通知第三方审核机构,第三方审核机构将最终结果通知申请人。
按上图所示,510(k)申请人首先向510(k)第三方审核机构递交技术文件,第三方审核机构按照与FDA相同的标准审核完成后向FDA递交推荐意见,FDA审核之后做出最终决定并通知第三方审核机构,第三方审核机构将最终结果通知申请人。02什么是EUA第三方审核
在本指南中,FDA除了提到510(K)第三方审核机构,还提到了EUA第三方审核。该机构旨在支持FDA保护和促进公众健康的使命,使该机构能够“激增”或*扩大其审核资源,用于审核与医疗器械相关的EUA请求。
根据《联邦食品、药品和化妆品法》*564条,FDA可在卫生与公众服务部长(HHS)发布紧急情况或威胁声明,证明授权紧急使用是正当的(“EUA声明”)后,授权在某些紧急情况下紧急使用未经批准的产品。

03哪些器械可申请510(K)第三方审核
目前,符合510(K)第三方审核路径审核要求的器械主要包括 I 类及部分 II 类器械,高风险III类及较为复杂的II类器械不在510(k)第三方审核机构的审核范围内。
FDA会动态更新产品列表,使FDA始终可将审查资源集中于高风险及复杂器械,同时保持对第三方审核项目的高度信任,现如今涵盖的器械大类如下:

在向510(K)第三方审核机构提出申请之前,制造商应先浏览FDA官网上的第三方审核的器械清单及不同第三方审核机构所能审核的器械目录。
04哪些机构510(K)第三方审核资质
根据《联邦食品、药品和化妆品法》*523条,当经认可的第三方实质上不符合相关规定时,FDA可以在提供通知后暂停或撤销对任何第三方审查组织的认可。
上海角宿企业管理咨询有限公司专注于FDA510(K),N95认证,TGA注册,欧代注册,欧洲自由销售证明,MDR认证,ISO13485认证,SFDA注册,FDA注册等